
Пошарова топографія. Шкіра тильної поверхні пальців тонка, підошвової — щільна, особливо в ділянці проксимальної фаланги. Підшкірна жирова клітковина на тильній поверхні пальців розвинена слабко, на підошвовій пронизана сполучнотканинними перетинками та має виражену комірчасту будову. Тильний апоневроз пальців укріплений сухожилками м'язів-розгиначів які кріпляться до фаланг пальців. З підошвового боку сухожилки м'... Читати далі...
14:44 Патологія органел цитоплазми |
Ендоплазматична сітка (лат. reticulum endoplasmicum, EC) — це сукупність канальців, мішечків і цистерн, утворених мембранами, що безпосередньо контактують з плазмолемою клітини та каріолемою. Розрізняють два типи EC: гранулярну та агранулярну. Перша представлена сукупністю канальців, мішечків і цистерн, до мембран яких з боку гіалоплазми прикріплені рибосоми. Функція гранулярної EC, крім біосинтезу білків, полягає в транспортуванні цих білків в інші ділянки клітини.
Агранулярна ендоплазматична сітка — це система розгалужених канальців, вакуолей і цистерн, на мембранах яких, на відміну від гранулярної ЕС, відсутні рибосоми. Функціями агранулярної (гладенької) ЕС є: участь у синтезі ліпідів, ліпопротеїдів, стероїдних гормонів, дезактивація різних шкідливих речовин та лікарських препаратів, синтез глікогену. Ступінь складності будови цієї органели пов'язаний з функціональною активністю клітини, в якій вона міститься. Зміни гранулярної і агранулярної ЕС можуть свідчити про пошкодження різних функцій клітини і проявляються гіперплазією і атрофією, дезагрегацією (дисоціацією) рибосом і полісом, утворенням аномальних рибосомально-пластинчастих комплексів. Гіперплазія гранулярної ЕС і рибосом, тобто збільшення їхньої кількості, на світлооптичному рівні проявляється підвищеною базо-філією цитоплазми, яка відображає об'ємну щільність рибосом і є показником інтенсивності білкового синтезу в клітині. Розширення елементів ЕС, що часто зустрічається при патологічних пошкодженнях і в різних екстремальних умовах, очевидно, можна вважати гіпертрофією цих органел тільки в тому випадку, коли цей феномен зумовлений відповідним приростом мембран, що їх утворюють. В іншому випадку може мати місце вакуолізація цистерн, яку навряд чи можна розглядати як прояв істинної гіпертрофії. Атрофія гранулярної ЕС— це зменшення її розмірів, з частковою або повною редукцією компонентів і втратою канальцями мембранозв'язаних рибосом. Усе це свідчить про зниження синтезу білка клітиною (білковий дефіцит при голоданні, хворобах печінки, старінні). Гіперплазія канальців агранулярної ендоплазматичної сітки пов'язана з посиленням метаболічної активності речовин ліпідної природи. Атрофія гладенької ендоплазматичної сітки виникає при гострій або хронічній дії на клітину різних токсичних речовин, а також при білковому голоданні. Таким чином, зміни ендоплазматичної сітки досить стереотипні: збільшення (при гіперфункції клітини) або зменшення (при гіпофункції клітини) кількості та розмірів канальців, концентричне розташування мембран, розширення канальців з появою в нихосміофільного вмісту (при токсичній дії), вакуолізація, фрагментація, зникнення рибосом, накопичення в канальцях фрагментів мембран, залишків клітинних органел (при дистрофії та некробіозі). Рибосоми розміщуються на мембранах ендоплазматичної сітки, а також у цитоплазмі у вигляді полісом. Кількість рибосом у різних клітинах і органах значно варіює, особливо багато їх у недиференційованих клітинах. При зменшенні синтезу білків кількість рибосом зменшується, при підвищенні, навпаки, збільшується. Зміни рибосом, як правило, поєднуються з патологією ендоплазматичної сітки. Комплекс Гольджі (лат. Complex Golgi, синоніми: внутрішній сітчастий апарат, пластинчастий комплекс) — мембранна органела загального призначення. Основними структурними компонентами його є скупчення сплющених цистерн, маленьких пухирців та великих вакуолей. Весь цей комплекс мембранних утворень називають диктіосомою, вона має дві поверхні (випукла проксимальна та вігнута дистальна). Функції комплексу Гольджі: накопичення, хімічна модифікація та дозрівання продуктів, синтезованих в елементах гранулярної ендоплазматичної сітки; синтез полісахаридів, глікопротеїдів, ряду ферментів та гормонів; участь в утворенні лізосом та пероксисом. Морфологічними проявами порушення діяльності пластинчастого комплексу і секретоутворення може бути гіпертрофія та атрофія. Гіпертрофія комплексу Гольджі, тобто збільшення кількості диктіосом за рахунок гіперплазії його мембран, секреторних гранул, везикул і вакуолей, є проявом підвищеного синтезу і секреції білків, гліколіпідів або полісахаридів. Продукти синтезу можуть накопичуватися в гіпертрофованих диктіосомах комплексу Гольджі в результаті порушення секреції та виведення їх, що може супроводжуватись пошкодженням канальців. Приклад — накопичення жовчі в пластинчастому комплексі гепатоцитів при холестазі. Атрофія комплексу Гольджі, тобто зменшення його розмірів з редукцією окремих компонентів, свідчить про зниження його функціональної активності. Однією з причин цього процесу є недостатність білкових запасів організму. У зв'язку з тим, що синтетична діяльність цієї органели тісно пов'язана з ендоплазматичною сіткою, другою причиною атрофії є порушення взаємодії пластинчастого комплексу з ендоплазматичною сіткою, тобто пошкодження внутрішньоклітинного конвеєра. Лізосоми (лат. lysosomae) — мембранні органели загального призначення. Вони представлені у вигляді електронно щільних утворень круглої форми діаметром 0,2-0,4 мкм, які містять біля 60 гідролітичних ферментів, основною функцією яких є розщеплення в клітині цілого ряду речовин (внутрішньоклітинне травлення). На зовнішній поверхні їхніх мембран розташований розвинений рецепторний апарат.
Установлено, що лізосоми здатні активно переміщуватись у клітині. Ферментами лізосом є білки, які синтезуються в гранулярній ендоплазматичній сітці, і за допомогою транспортних везикул переносяться до комплексу Гольджі, де приєднується вуглеводний компонент. Від зрілої поверхні комплексу у вигляді мембранних пухирців, наповнених ферментами, відділяються первинні лізосоми. Крім первинних лізосом, у клітині можуть бути і вторинні лізосоми, або фагосоми. Вони утворюються в процесі фагоцитозу.
При взаємодії первинної лізосоми з піноцитозними пухирцями (фагосома з рідким вмістом) утворюється інший тип вторинної лізосоми, яка називається мультивезикулярним тільцем. Вторинна лізосома з залишками непереварених речовин отримала назву залишкового тільця, яке виводиться з клітини шляхом экзоцитозу. Лізосоми можуть зливатися з пошкодженими фрагментами клітини (мітохондріями, ендоплазматичною сіткою та ін.), утворюючи аутофагосоми. Аутофагічна діяльність лізосом відіграє важливу роль при фізіологічній регенерації окремих внутрішньоклітинних структур, в процесах ембріогенезу, морфогенезу, та диференціювання клітин. Тривале перебування аутофагосом у клітинах може призводити до накопичення пігменту — ліпофусцину, який інакше називають "пігментом старіння" і виявляють у нервових клітинах мозку, печінки та м'язових волокнах. Група ліпопігментів включає також цероїд, який зустрічається в макрофагах. Зазначені пігменти мають у своєму складі -білки та важкорозчинні ліпіди. Значення лізосом у патології різноманітне, і важко уявити патологічний процес, в якому вони б не брали участі. Остання може бути двоякою. По-перше, вони відіграють важливу роль у розвитку таких патологічних процесів, як запалення, некроз, гіпоксія, голод та інші, де участь лізосом має вторинний характер.
По-друге, може мати місце пошкодження структури самих лізосом, що супроводжується розвитком патологічних процесів. У цих випадках пошкодження лізосом є первинним і розглядається як пусковий механізм у розвитку захворювання.
До патології лізосомальних мембран можуть призводити різні мембранотропні речовини екзогенного та ендогенного походження. Проникність мембран лізосом значно підвищується при гіпоксії, дії токсичних речовин та лікарських препаратів, змінах кислотно-лужної рівноваги, після травм та оперативних втручань, з!мін гормонального статусу організму, дії радіації, при деяких захворюваннях суглобів, атеросклерозі, інфекційних захворюваннях.
Серед патологічних станів, які пов'язані з пошкодженням лізосомальних мембран, найбільш вивчені хвороби суглобів. У патогенезі цих хвороб важлива роль належить комплексу ферментів, які звільнюються з лізосом. В основному, це кислі гідролази і, перш за все, протеази типу катепсину Д, які здатні руйнувати глікопротеїдні структури хряща, а також глюкуронідази, колагенази і катіонні білки. Речовини, що стабілізують лізосомальні мембрани (до них належать і саліцилати), значно зменшують ступінь запалення.
На особливу увагу заслуговує дія лікарських препаратів на лізосомальні ферменти пухлинних клітин, що можна розглядати як один із можливих підходів до цілеспрямованого використання хіміотерапевтичних засобів. Так, при дії цитотоксину або його комбінацій з вітаміном А виявлено збільшення кількості лізосом у клітинах, активація кислої фосфатази та вихід її в цитоплазму, що характеризується зростанням дистрофічних змін у клітинах пухлини, затримує їхній ріст. Відсутність у людини одного або кількох лізосомальних ферментів проявляється генетично зумовленими тяжкими спадковими захворюваннями — хвороби накопичення (тезаурисмози). Група спадкових лізосомних ензимопатій досить велика. Особливо чітко вона представлена серед гл ікогенозів (хвороба Помпе), гангліозидозів (хвороба Тея-Сакса, Сандхофа, ювенільний гангліозидоз), гепатозів (хвороба Дабина-Джонсона), ожиріння (недостатність ліпаз адипозоцитів). Друга група спадкових захворювань пов'язана з пошкодженням функції лізосом, що призводить до порушення мембранних взаємодій між органелами і утворень велетенських органел, у тому числі велетенських лізосом. Ця група захворювань невелика: синдром Чедіака-Хігасі, або так звана циклічна нейтропенія. Пероксисоми (peroxysoma) — мембранні органели спеціального призначення. Це округлої форми структури діаметром 0,2-0,5 мкм. Вони мають гранулярний матрикс, у центрі якого містяться кристало-подобні структури (серцевина, нуклеоїд). Гістохімічним маркером пероксисом є фермент каталаза, який розкладає токсичний для клітини перекис водню, що утворюється в процесі перекисного окислення. Основна функція пероксисом—утилізація хімічно активного атомарного кисню, а також розщеплення етилового спирту, сечової кислоти, регуляція обміну ліпідів. Зміни кількості та структури пероксисом зустрічаються при багатьох захворюваннях. Вони можуть бути первинними, що дозволяє говорити про "пероксисомні хвороби", клінічним проявом яких є каталазна недостатність, і вторинними, які відображають пошкодження оксидазно-каталазної активності клітини і пов'язані зі зміною числа цих органел. Збільшення кількості пероксисом і підвищення каталазної активності в гепатоцитах і нефроцитах можна викликати в експерименті за допомогою окремих лікарських препаратів, що мають гіполіпопротеїнемічну дію, а в кардіоміоцитах — при тривалому прийомі етанолу. У людини відзначається підвищення числа пероксисом в гепатоцитах при вірусному гепатиті, лептоспірозі. Зменшення числа пероксисом, особливо в гепатоцитах, викликають в експерименті за допомогою речовин, що гальмують синтез каталаз. У людини зменшення кількості пероксисом і зниження синтезу їхніх ферментів спостерігається в печінці при запаленні, а також при пухлинному рості. Значні дефекти пероксисомної системи або руйнування пероксисом спостерігаються при гіперліпідемії і гіперхолестеринемії. У цих випадках руйнування пероксисом відбувається в результаті аутолізу або аутофагії. Нуклеоїди пероксисом руйнуються також при введені тваринам речовин, які зменшують ліпідемію або після опромінення. У людини при деяких захворюваннях (гепатоцеребральна дистрофія) відбувається деградація нуклеоїдів пероксисом, при інших (ідеопатичний холестаз) — новоутворення нуклеоїдів в пероксисомах. Пероксисомний матрикс руйнується у тварин при введені їм інгібіторів синтезу катал аз. У людини руйнування матриксу пероксисом спостерігають при ішемічному некрозі та вірусному гепатиті.
Пероксисомні хвороби пов'язані з метаболічними розладами і є спадковими. Наприклад, акаталаземія, при якій активність каталази в печінці та інших органах дуже низька, як результат зниження її термостабільності. Єдиний клінічний синдром цього захворювання — виразки порожнини рота. Цереброгепаторенальний синдром Целлвегера характеризується відсутністю пероксисом в гепатоцитах.
Каталазна активність печінки у цих хворих складає приблизно 20 % норми. Результатом недостатності пероксисом при цьому синдромі є порушення синтезу жовчних кислот. Системна недостатність карнатину клінічно характеризується міопатією з періодичними пошкодженнями функцій печінки і головного мозку. Виражений дефіцит карнатину спостерігається в скелетних м'язах, де не відбувається окислення жирних кислот.
Мітохондрії (лат. mitochonriae) — мембранні органели загального призначення, напівавтономна структура клітини. Вона має власний генетичний матеріал (ДНК) і може ділитись самостійно, незалежно від поділу клітини. Мітохондрія — органела синтезу АТФ (аденозинтрифосфат), який відбувається в результаті процесів окислення органічних субстратів і фосфорування АДФ (аденоз- индифосфат). Мітохондрії мають подвійну мембрану: зовнішню та внутрішню. Остання відмежовує внутрішній вміст органел, її матрикс і утворює складки або вип'ячування, які називаються кристами. В клітинах різного типу розмір, форма та число крист широко варіюють. Наприклад, в мітохондріях гепатоцитів наявні короткі, паралельно розташовані кристи, в кардіоміоцитах вони можуть повністю перетинати мітохондрію, в епітеліоцитах наднирників та в інших органах, які виробляють стероїди, кристи мають форму трубочок. При деяких патологічних станах кристи можуть змінювати свою форму і переходити з трубчастих у пластинчасті структури або навпаки. Число крист у мітохондріях також відображає інтенсивність енергетичних процесів. У клітинах зі значними енергетичними затратами (наприклад, в м'язевих) мітохондріальні кристи численніші ніж, наприклад, в гепатоцитах.
При патологічних процесах зміни мітохондрій досить стереотипні, хоча деякі патологічні стани несуть специфічні ознаки пошкодження. Серед змін структури мітохондрій найбільше значення має їх набухання та конденсація матриксу. Подібні зміни можуть відображати функціональне напруження клітини в умовах гіпоксії. Як правило, вони носять зворотний характер, але якщо прогресують, то приводять до загибелі мітохондрій. У цих випадках до набухання органел приєднується ущільнення їх матриксу, деформація крист, втрата мітохондріальних гранул, гомогенізація матриксу і розрив зовнішньої мітохондріальної мембрани (мал.8).  Мітохондрії є досить лабільними структурами і першими реагують на зміни при гіперфункції клітини та її пошкодженнях.
При гіпертрофії розміри мітохондрій збільшуються, можуть утворюватись велетенські мітохондрії за рахунок збільшення розмірів однієї органели або злиття кількох. Такі органели часто мають кристалічні включення і спостерігаються в гепатоцитах при алкоголізмі. Збільшення числа мітохондрій (гіперплазія) відображає посилення процесів окислювального фосфорування, що характерно для клітин з активацією спеціалізованих функцій і спостерігається при проліферації або трансформації клітин, особливо після пошкодження тканини. Збільшення кількості мітохондрій характерне для злоякісних пухлин, зменшення — для регресивних процесів, зокрема, старіння клітин, їх атрофії. Зміни крист, як і самих мітохондрій, можуть стосуватись їхньої структури, розмірів, форми та числа. Деформація і агрегація крист є проявом зниження активності мітохондрій. Однією з важливих функцій мітохондрій є транспортування кальцію. Ультраструктурно цей процес характеризується відкладанням у мітохондріальному матриксі електроннощільних гранул, які, можливо, є місцем акумуляції двовалентних іонів. При деяких захворюваннях (ішемічна хвороба серця), синдромах (хронічна недостатність нирок) і патологічних станах (отруєння тіоацетатамідом, папаїном, йодоформом та ін.) клітини у відповідь на пошкодження нагромаджують у мітохондріальному матриксі велику кількість осміофільних гранул кальцію. При цьому, кальцифікація мітохондрій може мати зворотний характер або ж передувати некрозу клітини. Кальцифікація мітохондрій може бути пов'язана як із надлишковим надходженням кальцію в клітину, в зв'язку з первинним пошкодженням плазматичної мембрани, так і з первинним пошкодженням транспорту кальцію мітохондріями. У цьому випадку накопичення кальцію в мітохондріях "забирає" енергію АТФ і пошкоджує саму систему генерації енергії — мітохондрії. Первинне пошкодження мітохондріального транспорту кальцію зустрічається при хворобах скелетних м'язів (хвороба Люфта, синдром Кернса-Сайра). При цих захворюваннях мітохондрії, незважаючи на високий рівень ендогенного кальцію, можуть ще додатково накопичувати значну його кількість. У таких випадках можна говорити про "хвороби пошкодження" мітохондріального транспорту. Таким чином, до основних ознак пошкодження мітохондрій слід віднести: набухання мітохондрій, просвітлення матриксу, його вакуолізацію, вогнищеву гомогенізацію, деструкцію та фрагментацію крист, дифузну гомогенізацію мітохондрій, появу вакуолей та включень кальцію, руйнування зовнішньої мембрани мітохондрії. |
|
|
 |
Розділ: | Категорія: Топографічна анатомія нижньої кінцівки
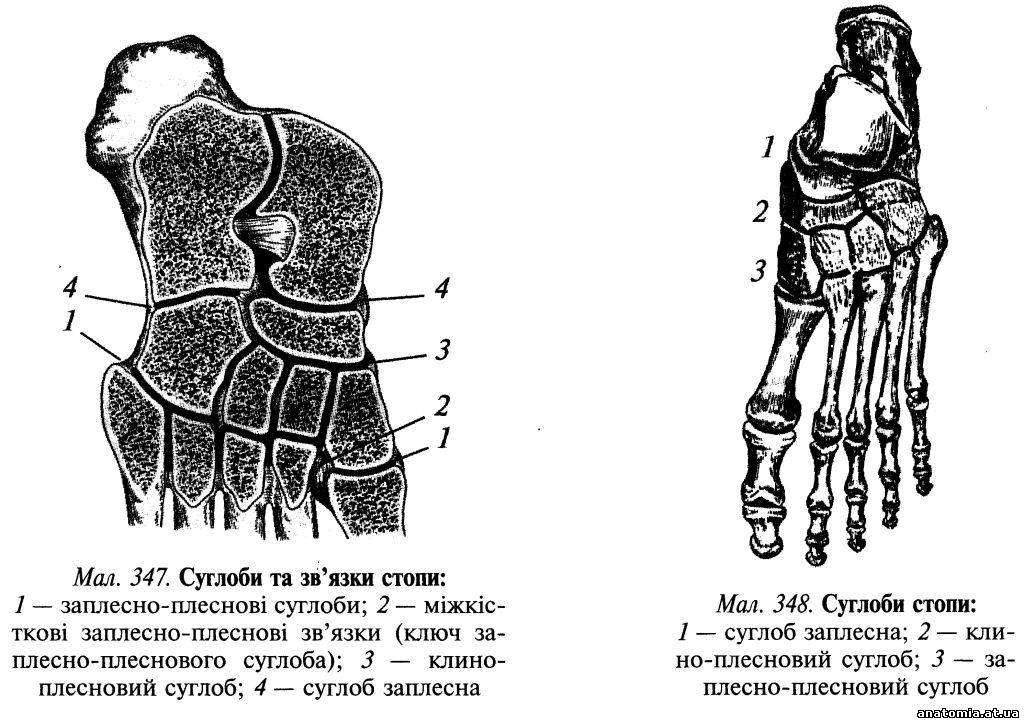 Пошарова топографія. Шкіра підошвової поверхні стопи товста та міцно зрощена з підлеглим підошвовим апоневрозом (aponeurosis plantaris) за допомогою великої кількості сполучнотканинних перегородок, які пронизують підшкірну жирову клітковину. Підшкірна жирова клітковина добре розвинена в ділянці п'яткового горба і головок плеснових кісток, де вона виконує роль амортизатора. Завдяки її вираженій комірковій будові нагнійні проц... Читати далі... |
Розділ: | Категорія: Топографічна анатомія нижньої кінцівки